一年一度的Oxford nanopore大會又來了,假如時間不夠,可以直接聽牛津孔洞的CEO James Clarke的演講Update from Oxford Nanopore Technologies,基本上就會是今年他們火力集中的部分,大部分會議中的演講,會慢慢在他們官方的youtube頻道釋出。
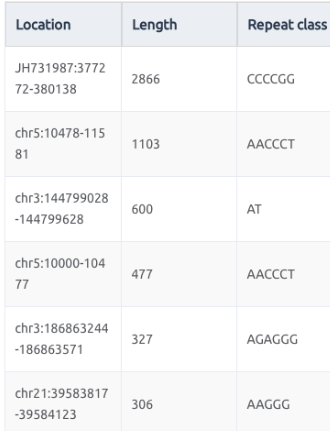
短串聯重複(Short Tandem Repeats,簡稱STRs)和微衛星(Microsatellites)這兩個在形容基因組結構的名詞,其實本質上是一樣的,但在體細胞談論遺傳時,都是以短串聯重複STRs為主,而在腫瘤領域在探討這類短片段重複的序列特性,則是在。它們都指的是一段由短的核苷酸序列(通常為2-6個核苷酸)構成的重複單元在基因組中連續出現的區域。
而這樣的重複片段在不同基因結構的區域,其實也有不少相關疾病被知道,比如上面圖片來自Hannan, A. J. (2018). Tandem repeats mediating genetic plasticity in health and disease. Nature Reviews Genetics, 19(5), 286-298.文章,便可以看到下面相關的疾病:
短串聯重複(Short Tandem Repeats,STRs)在基因組中具有高度變異性,與某些遺傳性疾病密切相關。以下是幾個與STRs相關的遺傳疾病範例:
肺動脈高壓(Pulmonary Arterial Hypertension,PAH):這是一種影響肺血管的疾病,與BMPR2基因中的STRs變異相關。該變異導致骨形成蛋白受體2(bone morphogenetic protein receptor type 2)的功能缺陷,使肺動脈壓力上升,進而引起心臟負擔加重。
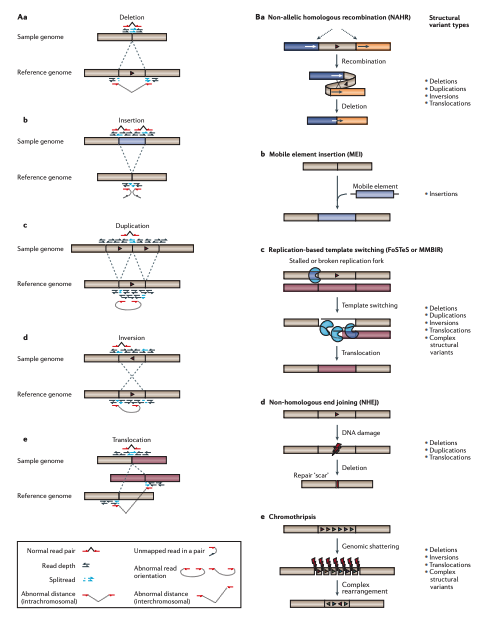
拷貝數變異(Copy Number Variants):在長一點的重複序列
上面的短串聯重複片段(STRs),既然叫做“短”,那麼就有在長一點點的重複序列特徵,那麼就會被歸類在所謂的拷貝數變異,歸類在更大範圍的結構變化(Structual Variation),換句話說,拷貝數變異(Copy Number Variants)涵蓋了一個相對廣泛的基因組片段大小範,從數百個核苷酸(bp)到數百萬個核苷酸(bp)不等。這些變異可以是重複(增加拷貝數)或缺失(減少拷貝數)。然而,CNVs的精確大小範圍會根據定義和檢測方法而有所不同。一些研究將CNVs定義為影響至少1,000個核苷酸(1 kb)的變異,而其他研究則將閾值設置為50,000個核苷酸(50 kb)或更大。隨著檢測技術的不斷進步,研究人員現在能夠在更細的尺度上檢測到更小的CNVs,進一步擴大了我們對這些變異的認識和研究範疇。
Redon, R., Ishikawa, S., Fitch, K.R., Feuk, L., Perry, G.H., Andrews, T.D., Fiegler, H., Shapero, M.H., Carson, A.R., Chen, W., Cho, E.K., Dallaire, S., Freeman, J.L., Gonzalez, J.R., Gratacos, M., Huang, J., Kalaitzopoulos, D., Komura, D., MacDonald, J.R., Marshall, C.R., Mei, R., Montgomery, L., Nishimura, K., Okamura, K., Shen, F., Somerville, M.J., Tchinda, J., Valsesia, A., Woodwark, C., Yang, F., Zhang, J., Zerjal, T., Zhang, J., Armengol, L., Conrad, D.F., Estivill, X., Tyler-Smith, C., Carter, N.P., Aburatani, H., Lee, C., Jones, K.W., Scherer, S.W., & Hurles, M.E. (2006). “Global variation in copy number in the human genome." Nature, 444(7118), 444-454.
Stankiewicz, P., & Lupski, J.R. (2010). “Structural Variation in the Human Genome and its Role in Disease." Annual Review of Medicine, 61, 437-455.
Weischenfeldt, J., Symmons, O., Spitz, F., & Korbel, J.O. (2013). “Phenotypic Impact of Genomic Structural Variation: Insights from and for Human Disease." Nature Reviews Genetics, 14(2), 125-138.
Zarrei, M., MacDonald, J.R., Merico, D., & Scherer, S.W. (2015). “A Copy Number Variation Map of the Human Genome." Nature Reviews Genetics, 16(3), 172-183.
跟3D基因體學相關的文獻
Dekker, J., Marti-Renom, M. A., & Mirny, L. A. (2013). Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nature Reviews Genetics, 14(6), 390-403.
Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., … & Dekker, J. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science, 326(5950), 289-293.
Dixon, J. R., Selvaraj, S., Yue, F., Kim, A., Li, Y., Shen, Y., … & Ren, B. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485(7398), 376-380.
Rao, S. S., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., … & Aiden, E. L. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159(7), 1665-1680.
Bonev, B., & Cavalli, G. (2016). Organization and function of the 3D genome. Nature Reviews Genetics, 17(11), 661-678.
# Shell 說明
#!/bin/bash
# Slurm 指令
#SBATCH -A ACD110078 # Account name/project number
#SBATCH -J hello_world # Job name
#SBATCH -p test # Partiotion name
#SBATCH -n 24 # Number of MPI tasks (i.e. processes)
#SBATCH -c 1 # Number of cores per MPI task
#SBATCH -N 3 # Maximum number of nodes to be allocated
#SBATCH -o %j.out # Path to the standard output file
#SBATCH -e %j.err # Path to the standard error ouput file
#程式與指令
module load compiler/intel/2020u4 IntelMPI/2020
mpiexec.hydra -bootstrap slurm -n 24 /home/user/bin/intel-hello